Орфанные заболевания — это малоисследованные состояния, специфические методы лечения которых пока неизвестны. Часто люди с такими проблемами чувствуют себя отринутыми и «потерянными» в мире здравоохранения. Разбираемся, почему так происходит.
Павлику, сыну Снежаны Митиной, было всего три года, когда ему поставили диагноз мукополисахаридоз II типа, или синдром Хантера. «Мы только открыли дверь, а врач уже говорит: „Какой прекрасный Хантер!“ — рассказывает Снежана теперь. — С первой частью я согласилась, а вот кто такой Хантер, мне тогда было неизвестно».
В детском саду мальчик был самым высоким в своей группе, читал стихи про бычка, который идет и качается. Поверить, что совсем скоро он перестанет ходить и говорить, казалось невозможным.
Но в четыре из-за гиперактивности Павлика исключат из детского сада, в шесть — из детского сада для инвалидов: перестанет усваивать программу. У него изменятся внешность и характер.
Терапии, способной корректировать часть симптомов, вместе с другими родителями Снежане придется добиваться самой. Элапразу — единственный препарат, способный помочь ее сыну, зарегистрируют в России не сразу и именно ее трудами.
Синдром Хантера — это одна из форм мукополисахаридоза, генетическое заболевание, возникающее в результате дефицита ряда ферментов, расщепляющих продукты обмена. Этот дефицит приводит к накоплению белково‑углеводных комплексов и жиров в клетках. В результате чего происходит «самоотравление» организма и поражаются органы.
В России редким, или орфанным, считается заболевание, которое встречается у 1 человека из 10 тысяч или реже. Именно из-за редкости такие болезни сложно диагностировать и лечить. Тот синдром, что у Паши, встречается у одного ребенка на 132 000 родившихся младенцев.
Сейчас Снежана помогает больным мукополисахаридозом как президент МБОО «Хантер-синдром»: рассказывает, как получить лекарство, через благотворительные фонды находит памперсы, инвалидные коляски и ортопедическую обувь.
Право выбора
Большинство редких болезней хронические. То есть неизлечимые. Почти все приводят к инвалидизации и смерти. Орфанные препараты, если терапия от конкретного недуга все-таки существует, как правило, не излечивают болезнь полностью, а лишь снимают тяжесть симптомов и приниматься должны на протяжении всей жизни.
«С первой частью я согласилась, а вот кто такой Хантер, мне тогда было неизвестно»
Лечение двадцати четырех самых тяжелых и дорогих орфанных заболеваний финансируется государством. Если диагноз входит в знаменитый список «12 нозологий», то лекарство закупают за счет федерального бюджета, если в «Перечень 24» — за препараты платят регионы. В остальных случаях пациенты получают терапию как жизненно необходимую или по инвалидности.
Известно, что крайне важно начать лечение от мукополисахаридоза как можно раньше. Павлик получил первые свои медикаменты только в восемь. Сейчас ему девятнадцать, с восемнадцати он не говорит, хотя понимает человеческую речь.
«Конечно, мне часто пишут мамы, — признается Снежана: „Мы видели вашего ребенка в интернете. Он не разговаривает и ходит в памперсах. Зачем нужна такая жизнь?“. Но у меня короткий ответ. Что вы выберете: двенадцать лет ходить на кладбище или двенадцать лет целовать сына?».
За двенадцать лет терапии Павел не пропустил ни одного приема Элапразы. Хоть препарат и стоит около миллиона (!) рублей в неделю. Благо в Москве проблем с лечением нет.
Вопрос денег
У сына Натальи Буртаевой мукополисахаридоз IV типа, заболевание схожее, но ни в списке «12 нозологий», ни в «Перечне 24» его нет.
Даниилу уже шестнадцать. Его рост 97 сантиметров. Ходит с трудом: ноги и руки деформированы, внутренние органы тоже деформированы. С пятого класса Даня учится на дому. Нервная система при этом диагнозе не страдает, дети с ним отлично осваивают школьную программу. Но поврежденной оказывается опорно-двигательная система, страдает слух.
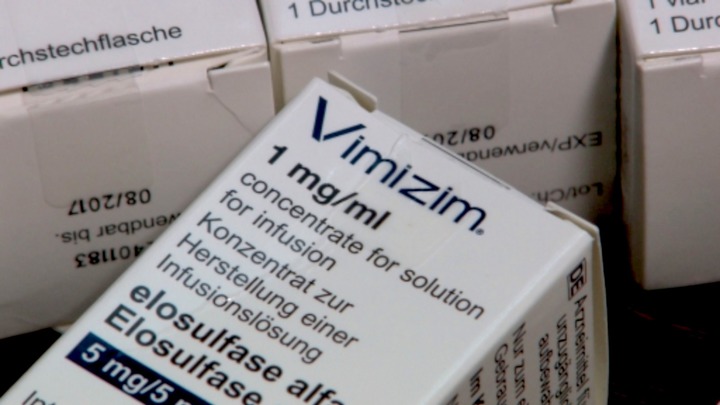
Препараты специальной ферментной заместительной терапии, положенной в таких случаях, существуют, но первое время были не зарегистрированы в России. Единственное лекарство, одобренное американским надзорным фармакологическим ведомством (FDA) в 2014 году, называется элосульфаза альфа — торговое название Вимизим (Vimizim).
Без Вимизима больные мукополисахаридозом IV типа умирают к двадцати годам. Когда новости о его разработке только появились в специализированных СМИ, Наталья было обрадовалась. Но стоимость его годового курса превышает 60 миллионов рублей.
Минздрав Ульяновской области ожидаемо отказался приобретать препарат. Наталья подала в суд и выиграла: первый в 2016 году, второй, когда министерство подало апелляцию, — в 2017. Ни письма в Росздравнадзор и Путину, ни сюжет на «Первом канале» не помогли: Даниил до сих пор не получает лечения.
«Мы не прячемся, — твердо сообщает Наталья корреспонденту. — Даня часто гуляет на коляске. Мы с ним ездили на море, а сегодня вообще особенный день: последний звонок. Он выйдет на сцену и получит аттестат зрелости.
Добавляет: «Он у меня обычно говорит, когда я переживаю, что люди его не примут: „Мам, ты, это, не переживай. Я привык, что на меня так смотрят“».
Более того, после школы Даниил планирует учиться дальше, признается мать, вздыхая: «Не знаю, сколько он будет жить». Врачи точно ответить на этот вопрос тоже не в силах, но очевидно, что без терапии Даня проживет недолго.
«Сделано в России»
Почему орфанные препараты стоят так дорого? Фармакологические компании разрабатывают их ради прибыли. Исследования нового лекарства стоят немало, а весьма небольшой тираж препарата призван не только возместить потраченные на него миллионы, но и «вернуться» с процентами.
Если в производстве инсулина в России заинтересованы миллионы, то «целевая аудитория» орфанных препаратов даже на такую большую страну, как наша, нередко не превышает пары десятков пациентов.
Во многих странах предусмотрена государственная поддержка исследователей, разработчиков и производителей орфанных препаратов: льготы, гранты и так далее. Но в России таких программ нет.
«Мам, ты, это, не переживай. Я привык, что на меня так смотрят»
Для того чтобы снизить издержки, власти предпочитают не субсидировать или договариваться с производителем, а искать оригинальным препаратам дешевые аналоги, дженерики. К сожалению, как убеждены некоторые эксперты, они нередко отличаются по эффективности.
Насте Катасоновой двадцать один, и она не знает, сколько раз лежала в больнице: до восьми лет — раз в год, до четырнадцати — по два раза, потом — по три, сейчас — госпитализироваться приходится чуть ли не каждый месяц.
Говорит, что в больнице ей спокойно: за твоим состоянием следят специалисты. Впрочем, и тут есть поводы для волнений. У Насти муковисцидоз, редкая болезнь легких, и недавно оригинальные антибиотики заменили отечественным дженериком.
Внешне люди с редкими заболеваниями часто не отличаются от здоровых. Однажды в школе кто-то из родителей одноклассников сказал, что девочке родственники купили инвалидность, дабы та не ходила на уроки. Одноклассники в ответ на слух объявили несчастной бойкот. Теперь она о муковисцидозе говорит открыто, чтобы не возникало кривотолков.
В ноябре Насте двадцать два. Между больницами и процедурами она профессионально фотографирует, ведет личный блог, помогает с соцсетями благотворительным организациям, занимается веганским магазином.
От побочных эффектов нового дженерика — у оригинального препарата их не было — состояние Анастасии ухудшилось. «Но не пить их нельзя, иначе — захлебнешься в мокроте», — объясняет пациентка.
«Каждое утро для меня — испытание, — рассказывает она. — Мокрота за ночь отлеживается в легких. Встаю. Но не так красиво, как в фильмах, а с кашлем. За день получается стакан густой зеленой жижи. Таблетки, ингаляция, если надо на работу, приходится просыпаться за два часа до выхода». Днем опять ингаляции. Перед сном питание: через гастростому. Это специальную трубка, которая устанавливается в отверстие на животе и ведет прямо в желудок.
Екатерина Захарова, руководитель лаборатории наследственных болезней обмена веществ, председатель экспертного совета по редким болезням Всероссийского общества орфанных заболеваний, считает, что нужен активный диалог с разработчиками и производителями орфанных препаратов: «Необходимо выяснять, какие лекарства фармкомпании могут предложить бесплатно, какую минимальную стоимость могут установить. Кроме того, важно понять, какие препараты Россия может производить самостоятельно, чтобы обеспечить своих граждан: возможно, в рамках госзадания будет дешевле создать российское лекарство для небольшого числа пациентов».
Преступная забывчивость
Впрочем, не все орфанные препараты стоят миллионы. Вот только получить даже относительно недорогое лекарство нуждающимся в нем порой оказывается совсем не просто.
У Виктории Рыжковой редкое генетическое заболевание. Синдром Вильсона-Коновалова, или гепатоцеребральная дистрофия.
Медь при этой болезни не выводится из организма, а проблемы с нарушением ее обмена ведут к накоплению элемента в нервной системе, почечной, печеночной тканях и роговице, что оборачивается токсическим повреждением указанных органов.
Заболевание бывает как врожденным, так и приобретенным. Болезнь у Виктории проявилась после неудачной беременности: сначала Вике парализовало пол-лица, потом появились проблемы с глотанием, передвижением, тремор рук. Врачи сперва списали все на наркоз, невроз и лишний вес. И только через полгода третий по счету невролог догадался отправить женщину на МРТ.
«Когда я пришла за результатами, там уже собралось все отделение, говорят: „Девушка, у вас токсическое поражение мозга, вы что, работаете с токсинами?“ — вспоминает теперь Вика.
Она не работала с токсинами, Виктория трудилась в «Ночлежке», помогала бездомным.
Только после МРТ врачи смогли поставить правильный диагноз. «Знакомые теперь не жалеют меня, а подшучивают, что у меня в голове стратегический металл, потому что медь применяют в оборонной промышленности», — иронизирует Вика.
Упаковка Купренила, препарата, убирающего лишнюю медь, стоит около двух с половиной тысяч рублей. Одной хватает на три месяца (некоторым нужно больше: три, пять, восемь, десять в месяц — в зависимости от сложности случая).
Стоимость Купренила для регионального бюджета относительно невысока: на одного больного ценник составляет максимум 300 000 в год, но регионы регулярно «забывают» его закупить — и приобретать необходимые медикаменты пациентам нередко приходится самостоятельно.
В 2016 году Купренил вовсе исчез из российских аптек. Израильская компания «Тева», которая производит препарат, закрывала линию по его производству на ремонт и предложила Минздраву РФ заранее увеличить объем закупок. Минздрав отказался. В итоге без препарата остались сотни пациентов по всей стране.
Чтобы раздобыть лекарство, необходимое для восстановления, Виктории пришлось покупать таблетки в Украине.
«У нас тогда с Митей Алешковским, соучредителем благотворительного фонда «Нужна помощь», даже был план, что, если меня задержат на границе, а я везла в три раза больше разрешенной нормы, он поднимет скандал в СМИ», — вспоминает теперь женщина.
Но обошлось. Въехать и выехать из страны удалось без особых потерь. Несмотря на обострение отношений между государствами и понятное в условиях конфликта недоверие таможенников.
Оптимистичный диагноз
Проблемы с диагностикой, как те, с которыми столкнулась Виктория, — явление распространенное. Кроме широко известных орфанных заболеваний, вроде гемофилии (царской болезни), несовершенного остеогенеза (хрустальной болезни), гипофизарного нанизма (карликовости), среди них есть и те, о которых широкой публике практически неизвестно.
Более того, значительное их число плохо изучено, а их симптомы незнакомы даже врачам.
В результате специалисты, к которым обращаются пациенты, как правило, бывают не в состоянии поставить им правильный диагноз с первого раза и тем более оперативно подобрать лечение.
«Пациенты приходят к врачам в регионах с выписками, чтобы получить лекарства, а те говорят: „Кто это вам назначил? Нельзя!“» — рассказывает Ирина Мясникова, председатель правления Всероссийского общества орфанных заболеваний (ВООЗ).
Надежда Титаренко — молодая блогерка и активистка из Смоленска — одна из тех, кто столкнулся с подобной проблемой. У нее спинально-мышечная атрофия (СМА) Кугельберга-Веландера — тяжелое заболевание, при котором вследствие генных мутаций оказываются повреждены части спинного мозга.
Cпециалисты, к которым обращаются пациенты, как правило, бывают не в состоянии поставить им правильный диагноз с первого раза.
Верный диагноз женщина узнала лишь в 2017 году, а до этого в течение двадцати пяти лет в ее карте значилась болезнь Верднига-Гоффмана. Врачи не верили, что Надя доживет до пяти лет, и даже после того, как прогнозы не подтвердились, отказывались назначать ей физиотерапию: «Всю жизнь врачи не хотели меня лечить, не хотели назначать санаторно-курортное лечение, не говоря уже о занятиях лечебной физкультурой. Считалось что это бессмысленно при „смертельном“ диагнозе».
Недавно Надя сделала операцию по выпрямлению позвоночника. В России их не делают, так что пришлось поехать в Финляндию. Подписчики ее блога помогли собрать деньги.
«Восемь часов — и я проснулась с новым телом. Пришлось заново учиться глотать, держать ложку и краситься, потому что меня вытянули на целых 15 сантиметров», — рассказывает теперь Надя. Сейчас, чтобы свободно гулять по родному городу, она собирает деньги на электроколяску «мечты» и жалеет, что из-за врачебной ошибки потеряла так много времени.
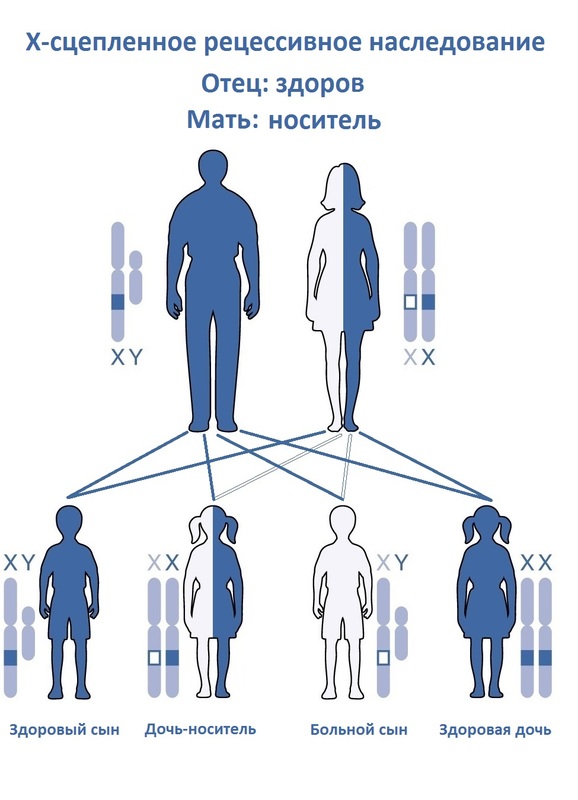
80 % орфанных заболеваний — генетические и передаются по аутосомно-рецессивному типу.
По мнению Екатерины Захаровой, чтобы избежать этой и многих других проблем, имеет смысл расширить скрининг новорожденных на редкие болезни: «В США новорожденных на пятьдесят разных заболеваний тестируют, а у нас только на пять. Около двух тысяч детей рождаются с наследственными нарушениями обмена веществ, иммунодефицитами, при ранней диагностике которых и своевременном начале лечения смертельного исхода можно избежать».
Всего известных редких заболеваний около 7 тысяч. И по каждому найдется не одна, а с десяток историй, подобных тем, что мы уже рассказали. Правда в том, что, даже если государство наладит упомянутый скрининг, вместе с выявлением новых пациентов ему придется решить и все названные выше проблемы по каждому из тысяч диагнозов. И пусть по отдельным пунктам ситуацию время от времени удается поправить, в том числе не без самоотверженного труда матерей, активистов и волонтеров, как скоро по всем из них удастся добиться изменений — вопрос открытый. Оптимистичный ответ на него звучит так: «К сожалению, нескоро». Пессимистичный же ни родственники, ни сами пациенты предпочитают не произносить лишний раз вслух.